
Transposição de doses de estudos pré-clínicos para humanos
A dose inicial em humanos não nasce de um palpite: ela é calculada com base em estudos pré-clínicos, alometria e avaliação comparativa entre espécies. Diferenças no metabolismo e no trato gastrointestinal fazem toda a diferença. Saiba como esse processo garante segurança e previsibilidade nos ensaios clínicos.
BLOG CIENP
Sara Tolouei, PhD; Fabiana C. V. Giusti, PhD e João B. Calixto, PhD
12/9/20255 min read
1. Introdução
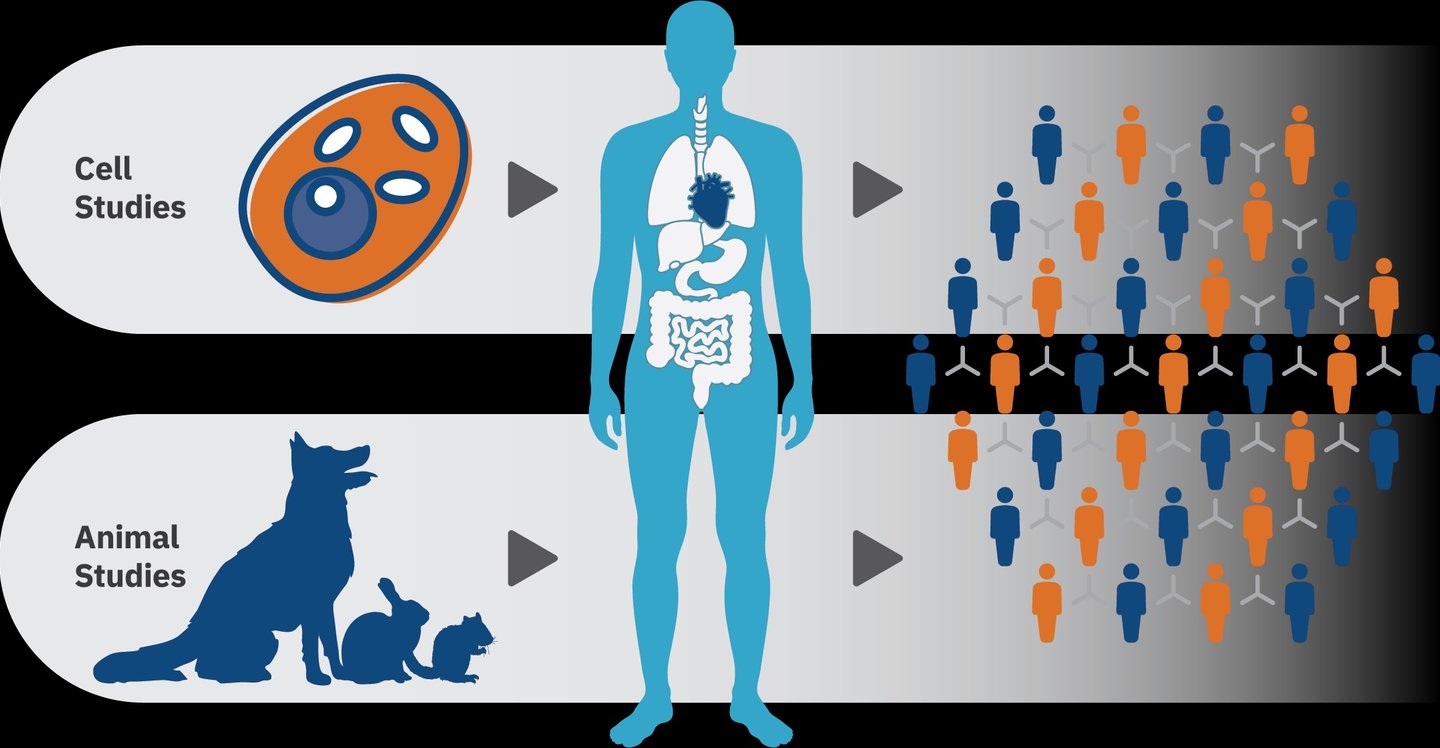
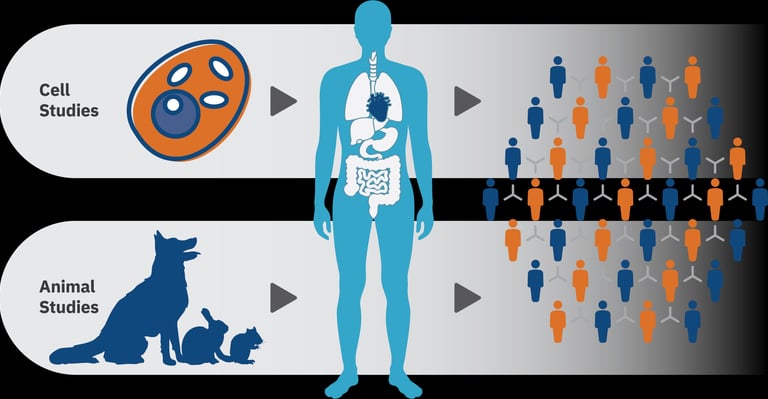
O desenvolvimento de um novo medicamento é um processo longo, complexo e altamente regulado, que exige a integração equilibrada entre inovação científica, eficácia terapêutica e garantia de segurança. A partir da identificação de uma molécula ou composto bioativo com potencial farmacológico, inicia-se uma etapa crítica: a investigação pré-clínica. Nessa fase, realizam-se estudos pré-clínicos in vitro e in vivo, especialmente em modelos animais, para caracterizar o comportamento do candidato a fármaco no organismo vivo. Avaliam-se parâmetros farmacocinéticos (absorção, distribuição, metabolismo e excreção), farmacodinâmicos (mecanismos de ação e efeitos biológicos) e toxicológicos, para garantir a segurança, com o objetivo de identificar riscos, compreender possíveis efeitos adversos e definir margens seguras de exposição.
Essas avaliações pré-clínicas são indispensáveis para fundamentar decisões regulatórias e científicas sobre a progressão do composto para os estudos clínicos em humanos. Elas permitem prever cenários de segurança, estabelecer doses iniciais para ensaios clínicos e reduzir incertezas na transição para a pesquisa clínica, contribuindo para o desenvolvimento responsável e robusto de novos medicamentos.
Um ponto central nesse processo é definir com segurança a dose inicial em humanos (First-in-Human, FIH). A escolha dessa dose deve ser suficientemente baixa para garantir segurança, mas também adequada para possibilitar a detecção de efeitos farmacológicos. A simples extrapolação aritmética da dose usada em animais não é adequada, uma vez que diferenças interespécies podem alterar a absorção, o metabolismo e a toxicidade de forma significativa.
Nesse contexto, a alometria tem sido a abordagem mais amplamente utilizada para a transposição de doses de animais para a espécie humana. Ela permite considerar não apenas o peso corporal, mas também relações fisiológicas que variam de forma proporcional à superfície corporal e ao metabolismo basal.
2. Alometria e diferenças do trato gastrointestinal entre espécies
Conceito de alometria
A alometria descreve a relação entre tamanho corporal e parâmetros fisiológicos ou farmacocinéticos. Ela parte da observação de que funções metabólicas não aumentam de forma linear com o peso corporal, mas seguem leis de potência. Um exemplo clássico é a lei de Kleiber, segundo a qual a taxa metabólica basal é proporcional à massa corporal elevada a 3/4.
Com base nesses princípios, a extrapolação de doses de animais para humanos é feita ajustando-se a dose em mg/kg dos animais pela razão dos fatores Km (peso corporal / área de superfície corporal) fornecidos em guias regulatórios, como o da FDA.
Diferenças do trato gastrointestinal (TGI)
Embora a alometria seja útil, ela não captura todas as variações entre espécies, sobretudo quando o fármaco é administrado por via oral. O TGI apresenta diferenças críticas:
Área e tempo de absorção: o intestino humano é muito mais extenso do que o de roedores, resultando em maior tempo de trânsito e grande superfície de absorção. Isso pode favorecer a absorção de moléculas pouco solúveis.
pH gástrico e intestinal: ratos têm em geral pH estomacal mais ácido em jejum quando comparado aos humanos, enquanto cães apresentam valores mais próximos dos humanos. Fármacos com solubilidade dependente de pH terão absorção distinta em cada espécie analisada.
Esvaziamento gástrico: em roedores, o esvaziamento é rápido, podendo reduzir a dissolução de compostos. Em humanos e cães, o esvaziamento gástrico é mais lento, facilitando maior tempo de contato.
Microbiota intestinal: espécies diferentes possuem composições bacterianas únicas, o que pode influenciar a degradação enzimática de compostos administrados oralmente.
Transportadores e enzimas metabolizadoras: a expressão de P-glicoproteína (P-gp), CYP3A e outras enzimas intestinais varia entre espécies, influenciando significativamente a biodisponibilidade oral.
Essas especificidades demonstram que a extrapolação alométrica é uma etapa inicial, mas precisa ser complementada pelo conhecimento da farmacocinética comparativa e de estudos em diferentes espécies.
3. Exemplos práticos de cálculos alométricos
A seguir, são mostrados exemplos de cálculo de Dose Humana Equivalente (Human Equivalent Dose, HED) a partir de diferentes espécies, usando a metodologia baseada na superfície corporal (BSA). Os fatores de conversão (Km) são valores médios recomendados pela FDA:


Exemplo 1 – Transposição de camundongo para humano
HED (mg/kg) = NOAEL obtida na espécie animal (mg/kg) multiplicado por Km animal/Km humano
NOAEL em camundongo = 100 mg/kg
Km (camundongo) = 3
Km (humano) = 37
HED = 100 × 3/37 = 8,1 mg/kg
Para um adulto de 60 kg:
8,1 × 60 = 486 mg/dia
Aplicando um fator de segurança de 10:
486/10 = 48,6 mg/dia
Exemplo 2 – Transposição de primata não humano para humano
NOAEL em macaco = 15 mg/kg
Km (macaco) = 12
Km (humano) = 37
HED = 15 × 12/37 = 4,9 mg/kg
Para um adulto de 60 kg:
4,9 × 60 = 294 mg/dia
Aplicando um fator de segurança de 10:
294/10 = 29,4 mg/dia
Comparação entre espécies
Notar que, para a mesma dose em mg/kg, a HED varia bastante entre espécies. O uso de primatas ou cães, que apresentam fisiologia gastrointestinal e metabolismo mais próximos dos humanos, costuma gerar estimativas mais conservadoras do que as obtidas em roedores. Por isso, estudos de toxicidade regulatória frequentemente exigem pelo menos duas espécies, sendo uma delas não roedora.
4. Referências e guias regulatórios consultados
ICH–International Council for Harmonisation.
M3(R2): Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals. 2009. Disponível em: https://www.ema.europa.eu/en/ich-m3-r2-non-clinical-safety-studies-conduct-human-clinical-trials-pharmaceuticals-scientific-guideline
European Medicines Agency (EMA).
Guideline on strategies to identify and mitigate risks for first-in-human and early clinical trials with investigational medicinal products. 2017. Disponível em: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-strategies-identify-and-mitigate-risks-first-human-and-early-clinical-trials-investigational-medicinal-products-revision-1_en.pdf
Harold Boxenbaum and Clifford DiLea - First-Time-in-Human Dose Selection: Allometric Thoughts and Perspectives. J Clin Pharmacol 1995;35:957-966
Shannon Reagan-Shaw, Minakshi Nihal, and Nihal Ahmad. Dose translation from animal to human studies revisited. The FASEB Journal 660 Vol. 22 , 2007.
Anroop B. Nair, Shery Jacob, A simple practice guide for dose conversion between animals and human. Journal of Basic and Clinical Pharmacy. 7: 28, 2016. doi 10.4103/0976-0105.177703.
Reagan-Shaw, S., Nihal, M., & Ahmad, N. (2008).
Dose translation from animal to human studies revisited. FASEB Journal, 22(3), 659–661.DOI: https://doi.org/10.1096/fj.07-9574LSF.
Boxenbaum, H. (1982). Interspecies scaling, allometry, physiological time, and the ground plan of pharmacokinetics. Journal of Pharmacokinetics and biopharmaceutics, 10, 201–227. DOI: https://doi.org/10.1007/BF010621.
Endereço
Av. Luiz Boiteaux Piazza, 1302 - Sapiens Parque, Cachoeira do Bom Jesus, Florianópolis, Santa Catarina, Brasil.
Segunda a sexta-feira das 08:00 às 17:00 horas.
©2024 - Centro de Inovação e Ensaios Pré-Clínicos
Mapa do site
Serviços
+55 (48) 3332-8400
contato@cienp.org.br