
Gargalos para a Transposição de Estudos Pré-Clínicos para Estudos Clínicos (Fase I) no Brasil – Parte I.
O Brasil ainda enfrenta desafios significativos na transição dos estudos pré-clínicos para a fase clínica, devido aos altos custos, à longa duração e às reduzidas taxas de sucesso, que dificultam o avanço no desenvolvimento de novos medicamentos.
BLOG CIENP
Fabiana C. V. Giusti, PhD; Sara Tolouei, PhD e João B. Calixto, PhD
3/11/202510 min read
O desenvolvimento de medicamentos é um processo complexo e multifacetado, cujo objetivo é criar novas terapias eficazes e seguras para uso humano. Esse processo envolve diversas etapas principais, como a identificação de alvos, a descoberta e melhoramento da molécula candidata a medicamento, realização de estudos pré-clínicos de segurança e eficácia, ensaios clínicos, aprovação regulatória e vigilância pós-comercialização. Cada uma dessas fases enfrenta desafios significativos, incluindo a necessidade de equipes multidisciplinares com pesquisadores experientes da academia e da indústria farmacêutica. Além disso, o desenvolvimento de novos medicamentos envolve altos custos, cronogramas extensos e baixas taxas de sucesso. Em média, o investimento necessário para desenvolver um novo medicamento é estimado em centenas de milhões de dólares, e o processo pode levar de 6 a 10 anos para ser concluído. Mesmo já na fase de ensaio clínico, a taxa de sucesso de novos fármacos é inferior a 10%.
Vários fatores contribuem para essas dificuldades. Muitas doenças são complexas e multifatoriais, o que torna desafiador identificar tratamentos eficazes e seguros. O próprio projeto de desenvolvimento de medicamentos é intrincado, com múltiplos estágios nos quais contratempos podem interromper completamente o progresso. Adicionalmente, todas as etapas necessitam atender a padrões regulatórios elevados de segurança, eficácia e qualidade. Essas exigências adicionam tempo, riscos e custos ao processo. Para enfrentar esses desafios, os cientistas estão explorando ativamente tecnologias e metodologias inovadoras, como por exemplo a inteligência artificial (IA), que surge como uma força transformadora com potencial para revolucionar o campo.
O Brasil ainda enfrenta gargalos significativos na transposição de estudos pré-clínicos para a fase clínica. Um dos principais obstáculos históricos era a falta de infraestrutura nacional qualificada e capacitada para realizar estudos pré-clínicos que avaliem a eficácia e a segurança de novas moléculas ou extratos candidatos a medicamentos e vacinas, seguindo os princípios das Boas Práticas de Laboratório (BPL), exigidos por agências reguladoras nacional (ANVISA) e internacionais como a Food and Drug Administration (FDA) e European Medicines Agency (EMA). Essa lacuna limitava a capacidade do país de avançar no desenvolvimento de medicamentos inovadores para as etapas clínicas subsequentes.
Para superar esse gargalo, o Ministério da Saúde (MS) e o Ministério da Ciência, Tecnologia e Inovações (MCTI) idealizaram e financiaram a criação do Centro de Inovação e Ensaios Pré-Clínicos (CIEnP), localizado no Sapiens Parque, em Florianópolis (SC). Inaugurado em 2014, o CIEnP possui estrutura física e capacitação técnico/científica para realizar a maioria dos estudos pré-clínicos necessários ao desenvolvimento de medicamentos, incluindo pequenas moléculas, biológicos, fitoterápicos além de vacinas. No entanto, por questões legais, o CIEnP não realiza estudos em espécies animais não roedoras, como por exemplo, cães e primatas. Essas etapas específicas dos estudos pré-clínicos ainda precisam ser realizadas no exterior, o que representa um desafio adicional em termos de custos, logística e tempo.
A criação do CIEnP foi, sem dúvida, um avanço importante, mas ainda há muito a ser feito para consolidar a capacidade do Brasil em desenvolver internamente medicamentos inovadores. A superação desses gargalos exige investimentos contínuos em infraestrutura, formação de recursos humanos especializados, além de políticas públicas de longo prazo que apoiem a inovação e incentivem a colaboração entre academia, indústria e governo. Somente com esforços urgentes, bem coordenados e de longo prazo o país poderá reduzir sua dependência externa e fortalecer sua posição no cenário global de desenvolvimento farmacêutico.
Decorridos 10 anos após a inauguração do CIEnP, algumas empresas, universidades, Instituições de Ciência e Tecnologia (ICTs), conseguiram concluir com sucesso os estudos pré-clínicos de projetos de inovação em medicamentos e vacinas, atendendo os princípios da BPL e estão demandando por unidades de pesquisa clínica certificadas pela ANVISA para trabalhar de acordo com as boas práticas de pesquisa clínica para realizar estudos de fase I. Adicionalmente, esses projetos para iniciar a fase clínica I, necessitam de estreita parceria com a ANVISA para discutir os protocolos clínicos e, em especial, a confecção do Manual para Submissão de Dossiê de Desenvolvimento Clínico de Medicamento (DDCM).
Estudos clínicos de fase I
O estudo clínico de fase I tem por objetivo avaliar a segurança, os efeitos colaterais, a melhor dose de um novo candidato a medicamento. Ele também pode testar a melhor maneira de administrar um novo tratamento (por exemplo, por via oral, infusão na veia, etc) e como o tratamento afeta o organismo. As primeiras doses de uma molécula candidata a medicamento são geralmente administradas gradualmente, aumentando-se aos poucos, até determinar a dose máxima que não cause efeitos colaterais prejudiciais. Os ensaios clínicos de fase I geralmente incluem apenas um pequeno número de voluntários. Com exceção dos medicamentos destinados ao tratamento do câncer e de algumas doenças infecciosas, os estudos de fase I são geralmente conduzidos em voluntários saudáveis. Esses ensaios clínicos ocorrem ao longo de vários meses e envolvem um pequeno grupo de aproximadamente 20 a 100 participantes. Esses participantes recebem uma dose única "primeira em humanos" de um candidato a novo medicamento - internados em unidades de pesquisa clínica acreditadas pelas agências reguladoras para realizar pesquisa clínica - geralmente por via oral ou intravenosa, e são rigorosamente avaliados por pesquisadores clínicos treinados para avaliar sua absorção, eliminação e, principalmente a segurança. O principal objetivo do estudo de fase I é encontrar a dose mais elevada do novo tratamento que pode ser administrada com segurança, i.e., sem causar efeitos colaterais graves. Embora o candidato a medicamento tenha sido testado em laboratório em estudos com animais, os potenciais efeitos adversos somente podem ser conhecidos, com certeza, após a conclusão do estudo clinico de fase I.
Requisitos necessários para solicitar a autorização e aprovação para realização de estudo clínico de fase I
Iniciar um ensaio clínico de Fase I para um novo candidato a medicamento envolve vários desafios, que podem ser amplamente categorizados em exigências científicas, regulatórias, logísticas e éticas.
1. Aprovação regulatória
Como discutido nas Newsletters anteriores, nos Estados Unidos e na Europa as empresas farmacêuticas precisam encaminhar para a FDA e EMA solicitação formal de aprovação para “Novo Medicamento Sob Investigação” antes de iniciar um ensaio de Fase I. Este documento deve incluir dados pré-clínicos abrangentes, informações sobre a fabricação do Ingrediente Farmacêutico Ativo (IFA), acompanhado do protocolo de ensaio clínico que será empregado para avaliar a segurança e eficácia do novo candidato a medicamento. Ainda, é necessário desenhar um protocolo clínico detalhado de fase I com uma estratégia de escalonamento de dose segura e eficaz. A dose inicial deve ser baixa o suficiente para minimizar o risco, mas alta o suficiente para potencialmente mostrar um efeito farmacológico.
2. Requisitos de dados pré-clínicos
-Estudos de genotoxicidade, farmacologia de segurança e de toxicologia aguda e de doses repetidas: estudos de segurança pré-clínicos abrangentes e realizados de acordo com os princípios da BPL são necessários para estabelecer uma dose inicial segura para ser testada em humanos. Esses estudos devem demonstrar que o candidato a medicamento é seguro para uso inicial em humanos;
-Dados de eficácia, farmacológica e mecanismo de ação: dados farmacológicos adequados, incluindo mecanismo de ação, eficácia in vitro e em modelos animais e estudos de farmacocinética devem ser fornecidos para justificar os testes que serão realizados em humanos;
-Dados sobre a distribuição e excreção: informar dados sobre a absorção, distribuição, metabolismo e excreção da nova molécula candidata a medicamento;
-Fabricação do IFA e controle de qualidade em conformidade com as boas práticas de manufatura (GMP): a fabricação do candidato a medicamento deve seguir rigorosamente as diretrizes da GMP, garantindo a isenção de impurezas para assegurar a qualidade, consistência e segurança do produto. A ampliação da escala de produção do IFA para ensaios clínicos de Fase I é uma etapa crucial para atender aos requisitos regulatórios e garantir a segurança do produto.
O Controle de Manufatura Química (CMC, do inglês Chemical Manufacturing Control) desempenha papel essencial no desenvolvimento e na produção de substâncias farmacêuticas, conforme estabelecido pelas diretrizes da FDA. O CMC abrange um conjunto de atividades e controles destinados a garantir a qualidade, a consistência, a segurança e a eficácia do fármaco, desde a síntese do IFA até a formulação do produto final. As diretrizes da FDA e da International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) para o CMC são rigorosas e exigem que os processos de fabricação sejam robustos, bem caracterizados e validados, garantindo conformidade com os padrões regulatórios.
Os principais objetivos do CMC incluem:
• Garantia de qualidade: assegurar que a substância ativa e o produto final atendam aos padrões de pureza, potência e estabilidade;
• Consistência do processo: demonstrar que o processo de fabricação é controlado e reprodutível em escala comercial;
• Conformidade regulatória: atender aos requisitos das GMP e outras diretrizes internacionais, como as estabelecidas pelo ICH.
As diretrizes da FDA e ICH para o CMC impõem requisitos rigorosos às indústrias farmacêuticas, especialmente na identificação e caracterização química de novas moléculas destinadas à fase clínica I. Entre os principais desafios, destacam-se: i) garantir que o produto não contenha componentes desconhecidos ou impuros; ii) evitar que a molécula apresente estruturas químicas associadas a toxicidade conhecida ou potencialmente elevada; iii) assegurar a estabilidade química do produto ao longo de todo o programa de testes; iv) definir um perfil de impurezas adequado para avaliar potenciais riscos à saúde. Além disso, para que os estudos pré-clínicos sejam válidos na avaliação da segurança dos ensaios clínicos em humanos, os patrocinadores devem comprovar que o candidato a medicamento utilizado nos estudos clínicos mantém a mesma qualidade e composição do composto empregado nos testes pré-clínicos de toxicologia animal que sustentam a segurança do estudo clínico proposto.
O CMC é composto por vários elementos essenciais, que devem ser detalhados e documentados para submissão à FDA. A transição do laboratório para o piloto requer refinar a rota sintética para garantir a reprodutibilidade em escalas maiores. Alguns parâmetros críticos do processo como, por exemplo, temperatura, tempo de reação, velocidade de mistura podem interferir na qualidade do IFA e devem ser monitorados com cuidado. Os guias do ICH Q9 definem como avaliar o gerenciamento dos riscos, como impurezas, rendimentos, etc.
Considerações finais e desafios a serem superados
Embora o Brasil conte com diversos grupos de pesquisa clínica distribuídos em hospitais e regiões distintas do país, muitos dos quais possuem reconhecimento nacional e internacional para realizar estudos nas fases mais avançadas do desenvolvimento de novos medicamentos, o mesmo não pode ser dito em relação à fase clínica I. Essa etapa exige pesquisadores clínicos qualificados em pesquisa biomédica, denominados internacionalmente como Medical Doctor/PhD ou equivalente. Esse perfil profissional ainda é escasso no país. Esses especialistas são essenciais para estabelecer as conexões sólidas com as etapas anteriores do desenvolvimento de medicamentos, especialmente na discussão e interpretação dos estudos pré-clínicos e na elaboração de protocolos clínicos para realizar a fase I, fatores críticos para a translação bem-sucedida para o desenvolvimento clínico.
Outro ponto crucial é a necessidade de políticas públicas que incentivem e apoiem o surgimento de unidades de pesquisa clínica credenciadas pela ANVISA e FDA, capacitadas tanto técnica, quanto estruturalmente para realizar estudos de fase I, incluindo a internação de voluntários saudáveis. Essas unidades são fundamentais para consolidar a infraestrutura necessária para pesquisas clinicas iniciais de segurança e seleção de doses para avaliação clínica de novos fármacos.
Além disso, o Brasil precisa urgentemente investir na formação de recursos humanos em Química Medicinal e áreas relacionadas e no apoio ao desenvolvimento de indústrias químicas capazes de realizar o escalonamento e a produção de lotes piloto de IFAs, seguindo as diretrizes da GMP e os guidelines da FDA, EMA e da ICH. Sem essa capacitação, o país continuará dependente de IFAs importados, o que encarece e limita a produção local de medicamentos.
O Brasil pode se inspirar em exemplos bem-sucedidos de países como Coreia do Sul, China e Índia, que, há 30 anos, estavam em estágios tecnológicos semelhantes ou até inferiores ao nosso. Esses países investiram em políticas arrojadas e de longo prazo para desenvolver internamente medicamentos e vacinas, fortalecendo centros de pesquisa, universidades, indústrias e startups de biotecnologia (ver para mais detalhes a Newsletter de janeiro de 2025). Não há razão para que o Brasil não siga o mesmo caminho, criando estratégias robustas e ambiciosas para impulsionar a inovação na área farmacêutica.
Em função do fraco avanço obtido nas políticas de inovação em saúde no Brasil nas últimas 3-4 décadas, infelizmente, o tempo disponível, é limitado, e os desafios a serem vencidos são significativos. Manter-se no patamar atual diante das enormes transformações globais nesta área exigirá esforços e recursos públicos cada vez maiores para garantir o acesso da população a medicamentos e vacinas. Além disso, a dependência externa em medicamentos e insumos farmacêuticos pode atingir níveis insustentáveis, comprometendo seriamente o financiamento e a sustentabilidade do SUS e mesmo a soberania nacional. Portanto, é imperativo agir de forma estratégica e urgente para superar esses gargalos e garantir um futuro mais autônomo e inovador para a saúde pública no Brasil.
Voltaremos a este tema na próxima Newsletter para discutir outro grande gargalo ainda existente nesta área e que o Brasil precisa superar que é o desenvolvimento da formulação farmacêutica de um candidato a medicamento.
Referências:
-Melk A, Ernst CGJE, Saenger T, Degraeuwe E, Schimmer BB, Walle JV, Ažukaitis A, Prakken B, Campo EE, Bertero MG. Structured programs to train the next generation of clinician scientists. Nature Medicine. 2025;31:24–27.
-Food and Drug Administration (FDA). Q7 Good Manufacturing Practice Guidance for Active Pharmaceutical Ingredients – Guidance for Industry. [Internet]. Available from: https://www.fda.gov/media/71518/download
-Therapeutic Goods Administration (TGA). Guideline on Strategies to Identify and Mitigate Risks for First-in-Human Clinical Trials with Investigational Medicinal Products.
-European Medicines Agency (EMA). Guideline on the Requirements for Quality Documentation Concerning Biological Investigational Medicinal Products in Clinical Trials – Revision 2. [Internet]. Available from: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-requirements-quality-documentation-concerning-biological-investigational-medicinal-products-clinical-trials-revision-2_en.pdf
-Food and Drug Administration (FDA). Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-Characterized, Therapeutic, Biotechnology-Derived Products – Guidance for Industry. [Internet]. Available from: https://www.fda.gov/regulatory-information/search-fda-guidance-documents/content-and-format-investigational-new-drug-applications-inds-phase-1-studies-drugs-including-well
-Food and Drug Administration (FDA). CGMP for Phase 1 Investigational Drugs – Guidance for Industry. [Internet]. Available from: https://www.fda.gov/media/70975/download
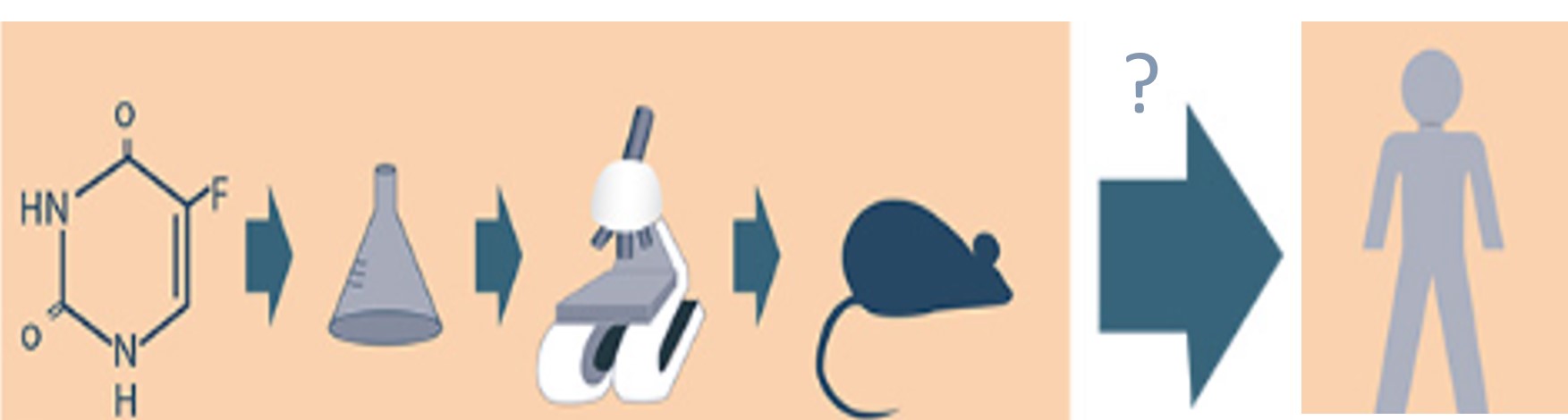
Fonte: Sindifar, 2018.
Endereço
Av. Luiz Boiteaux Piazza, 1302 - Sapiens Parque, Cachoeira do Bom Jesus, Florianópolis, Santa Catarina, Brasil.
Segunda a sexta-feira das 08:00 às 17:00 horas.
©2024 - Centro de Inovação e Ensaios Pré-Clínicos
Mapa do site
Serviços
+55 (48) 3332-8400
contato@cienp.org.br